
几何效应促进水滑石催化剂实现对羟醛缩合反应性能的提升
近日,《催化学报》在线发表了我课题组在固体碱催化领域的最新研究成果。该工作报道了不同离子半径元素的掺杂所产生的几何效应对催化活性位点的有效调控作用, 从而大大提升了羟醛缩合反应性能, 为制备新型优异的固体碱催化剂提供了思路和依据。论文第一作者为:王慧敏,论文共同通讯作者为:张欣,殷建军,卫敏。

背景介绍:近年来, 绿色发展观念深入人心。与液体碱相比, 固体碱催化剂由于其环境友好、腐蚀性小、易于回收等优点引起了科研工作者的广泛关注。但是, 在固体碱催化领域, 由于其体系的复杂性,如何对碱性位点进行调控从而使羟醛缩合反应获得优异性能仍然是一个巨大挑战。甲醛和异丁醛反应生成的产物羟基新戊醛是精细化工合成中一类非常重要的有机中间体, 广泛应用于药物、润滑油、聚酯树脂等化工产品生产,此反应已引起了人们极大的研究兴趣。目前为止,应用于该反应的固体碱催化剂催化活性较低,反应稳定性差,性能有待进一步提升。因此, 设计一类结构可调、性能优异的固体碱催化剂材料迫在眉睫。针对以上问题和挑战,本文利用水滑石材料结构特有的记忆效应制备了一系列掺杂镓、铟的钙铝水滑石催化剂, 并将其应用于甲醛与异丁醛缩合生成羟基新戊醛反应中,实现了对羟醛缩合反应的高效缩合;基于原位实验表征和理论计算手段,揭示了不同离子半径元素的掺杂所产生的几何效应对催化活性位点的有效调控作用, 从而大大提升了羟醛缩合反应性能, 为制备新型优异的固体碱催化剂提供了思路和依据。
研究思路:近年来的文献研究表明通过引入不同离子半径的金属阳离子进入LDHs主体晶格中所产生的几何效应对活性中心位点的微观结构起着至关重要的调节作用。一些金属离子作为结构助剂在氧化还原、加氢和缩合反应中已经被广泛应用。本文基于LDHs前体材料的元素组成可调变性和结构记忆效应,构筑了一系列Ga、In修饰的re-Ca4Al1-xMx-LDHs,并将其应用于甲醛和异丁醛羟醛缩合催化反应制备羟基新戊醛中;结果表明,re-Ca4A10.90Ga0.10-LDHs材料的催化性能最佳,对目标产物羟基新戊醛的产率可达到72.0%,明显优于未掺杂前re-Ca4A1-LDHs (60.1%)催化剂,并且与液体碱催化剂催化该反应所达到的性能(73.2%)基本接近,具有用量少、易分离、绿色环保等优势,具有潜在的实际应用前景。进一步采用XAFS、CDCl3-FTIR表征手段及DFT理论计算证明与单一的Al3+相比,Ga的修饰使得LDHs的晶格适度扩张,造成层板上Ca2+暴露更多的空s轨道,使其更容易与层间OH-配位形成7配位的Ca-OH位点,从而增加了弱碱性位点的浓度,使得催化性能显著提高。

图文解析
催化剂表征
首先,采用共沉淀法合成了Ca4Al1-xMx-LDHs(M=Ga、In)前体,随后,通过高温焙烧和水合复原过程制备得到re-Ca4Al1-xMx-LDHs(M=Ga、In)。XRD表明共沉淀法成功制备出具有良好晶型的纯相LDHs前体,由(003)特征衍射峰位置变化可以看出(Figure 1A),随着Ga和In的掺入,水滑石(003)特征衍射峰向低角度偏移,说明此时水滑石的层板间距变大,与Ga和In的引入对层板电荷密度及层板与层间阴离子的结合强度的改变有关。LDHs前体经过焙烧后,其特征衍射峰消失,出现CaO相的衍射峰,表明伴随着H2O和CO2的脱除其层状结构发生塌陷,此时LDHs前体已成功转化为Ca4Al1-xMx-MMOs结构。另外,没有观察到Ga2O3或者In2O3的衍射峰,说明其以无定型相存在于MMOs中(Figure 1B),随后在碱溶液中进行水合复原,氧化物重新恢复成水滑石的特征衍射峰(Figure 1C);另外可以发现,系列re-Ca4Al1-xMx-LDHs样品(003)衍射峰的位置均向高角度偏移至11o左右,这对应着OH-离子已成功的发生了离子交换反应而进入了水滑石的层间。
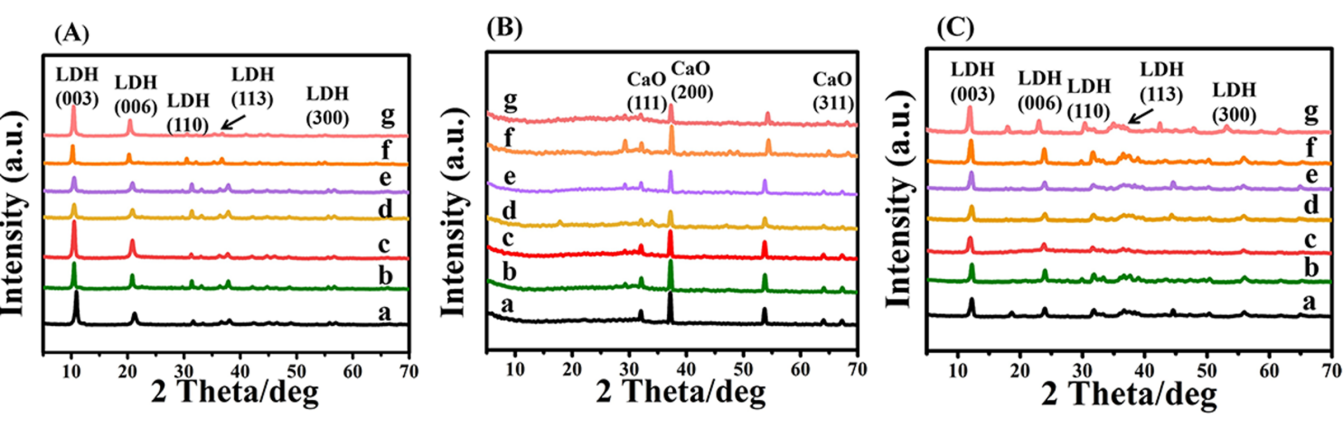
图1.XRD图谱 (A):Ca4Al1-xMx-LDHs(M=Ga、In)前体,(B): Ca4Al1-xMx (M=Ga、In)氧化物,(C):复原水滑石re-Ca4Al1-xMx(M=Ga、In)。其中,(a)为Ca4Al-LDHs, (b)为Ca4Al0.95Ga0.05-LDHs, (c)为Ca4Al0.90Ga0.10-LDHs, (d)为Ca4Al0.85Ga0.15-LDHs, (e)为Ca4Al0.95In0.05-LDHs, (f)为Ca4Al0.90In0.10-LDHs, (g)为Ca4Al0.85In0.15-LDHs
Ca4Al1-xMx (M=Ga、In)氧化物,(C):复原水滑石re-Ca4Al1-xMx(M=Ga、In)。其中,(a)为Ca4Al-LDHs, (b)为Ca4Al0.95Ga0.05-LDHs, (c)为Ca4Al0.90Ga0.10-LDHs, (d)为Ca4Al0.85Ga0.15-LDHs, (e)为Ca4Al0.95In0.05-LDHs, (f)为Ca4Al0.90In0.10-LDHs, (g)为Ca4Al0.85In0.15-LDHs
催化性能评价
随后经过对反应条件的筛选,我们在最佳条件下对七种水合复原后的re-Ca4Al1-xMx-LDHs样品进行了性能评价,其结果如图2A所示。对于系列re-Ca4Al1-xGax-LDHs催化剂,随着Ga3+/M3+原子比从0增加到0.15时,IBD的平衡转化率出现了先升高后降低的反应趋势。对比HPA的选择性,也表现出相似的趋势,其性能最大值(IBD转化率: 90.1%; HPA选择性: 80.5%)在re-Ca4Al0.90Ga0.10-LDHs催化剂上表现出来,在此催化剂上,HPA的最高产率为72.0%。另外,与其它催化剂进行比较,其系列re-Ca4Al1-xInx-LDHs催化剂的IBD转化率和HPA选择性均低于re-Ca4Al1-xGax-LDHs催化剂样品。此外,考虑到工业生产中的实际应用情况,进一步研究了re-Ca4Al0.90Ga0.10-LDHs催化剂的循环使用性,结果如图2B所示,将性能最优的re-Ca4Al0.90Ga0.10-LDHs进行多次重复利用,并测试其对该缩合反应的催化性能,结果表明,当对该催化剂进行6次的循环后,其反应的活性及对产物的选择性都能保持在一个稳定的水平,仅发生小部分的失活。
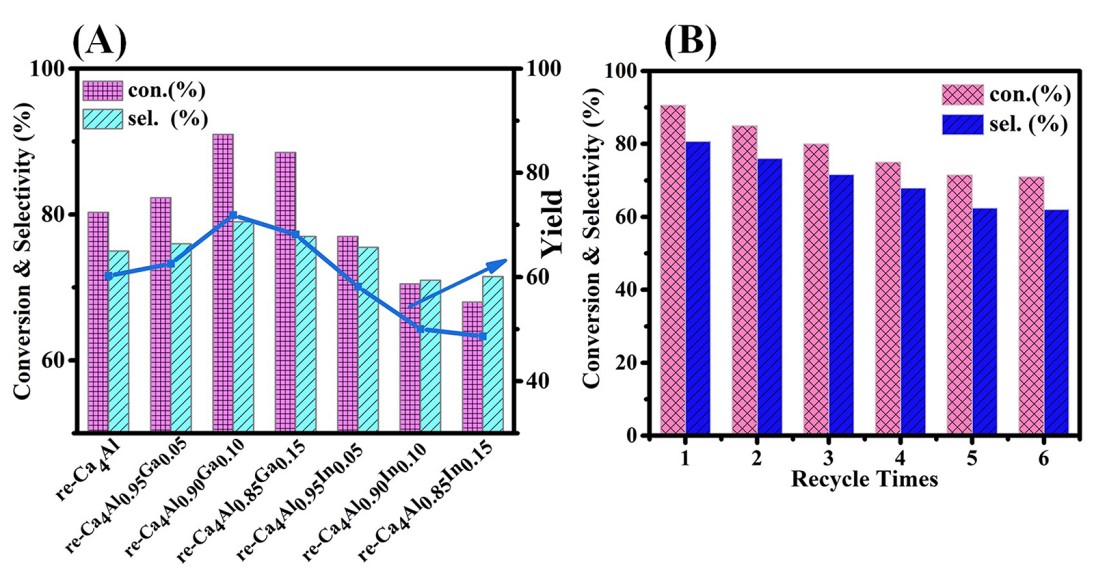
图2. (A) re-Ca4Al1-xMx-LDHs系列催化剂对IBD转化率和HPA选择性和产率的性能;(B)最优催化剂re-Ca4Al0.90Ga0.10-LDHs的循环稳定性(六次循环)
CDCl3-FTIR表面碱性位结构表征
为了探究系列re-Ca4Al1-xMx-LDHs催化材料表面碱性位点的分布状态,采用傅立叶变换的红外光谱简称为FTIR以及以氘代氯仿作为探针分子即CDCl3来进行原位吸附实验研究。将样品进行高温预处理后在真空环境下吸附CDCl3分子于催化剂表面,记录此时的红外谱图用以表征不同催化剂表面碱性位点的分布情况,图3为其原位红外结果。从图中看出,有一个宽泛并且不对称的吸收峰出现在了2180-2300 cm−1波数范围内。通过高斯拟合方法对其进行分峰处理,其中在2254 cm−1处和2214 cm−1处的红外振动峰可分别归属为弱碱性位点和中强碱性位点对CDCl3分子的吸附。基于前期对Ca4Al-LDHs的研究,证实了在复原的Ca4Al-LDHs固体碱材料催化体系中,弱碱性位点归属于Ca2+与OH-的7配位结构中的碱性氧位点,中强碱性位点则归属于物理吸附在水滑石层板表面上的羟基氧位点。此外,对于re-Ca4Al0.85Ga0.15-LDHs催化剂,除了在2254 cm−1和2214 cm−1观察到两个吸收峰外,在~2275 cm−1和~2264 cm−1波数处也出现了两个较小的红外吸收峰,分别归属于CDCl3气态和物理吸附在催化剂表面上的C-D键的伸缩振动。与re-Ca4Al-LDH相比,随着Ga3+/M3+原子比从0增加到0.15时,在弱碱性位点上关于CDCl3分子中C-D键的伸缩振动峰面积明显增大,其中,在re-Ca4Al0.90Ga0.10-LDHs催化剂上峰面积达到最大值。这说明Ga元素的掺杂增大了弱碱性位点(7-配位Ca-OH)的数量。而在re-Ca4Al1-xInx-LDHs催化剂中,则出现了与之相反的情况,其弱碱性位点的归一化峰面积随着In含量的增加而显著减小,而中强碱性位点的峰面积所占比例越来越大。此外,与re-Ca4Al-LDHs样品相比,re-Ca4Al1-xGax-LDHs和re-Ca4Al1-xInx-LDHs体系中弱碱性位点的吸收峰位置变化很微小,可以忽略不计。
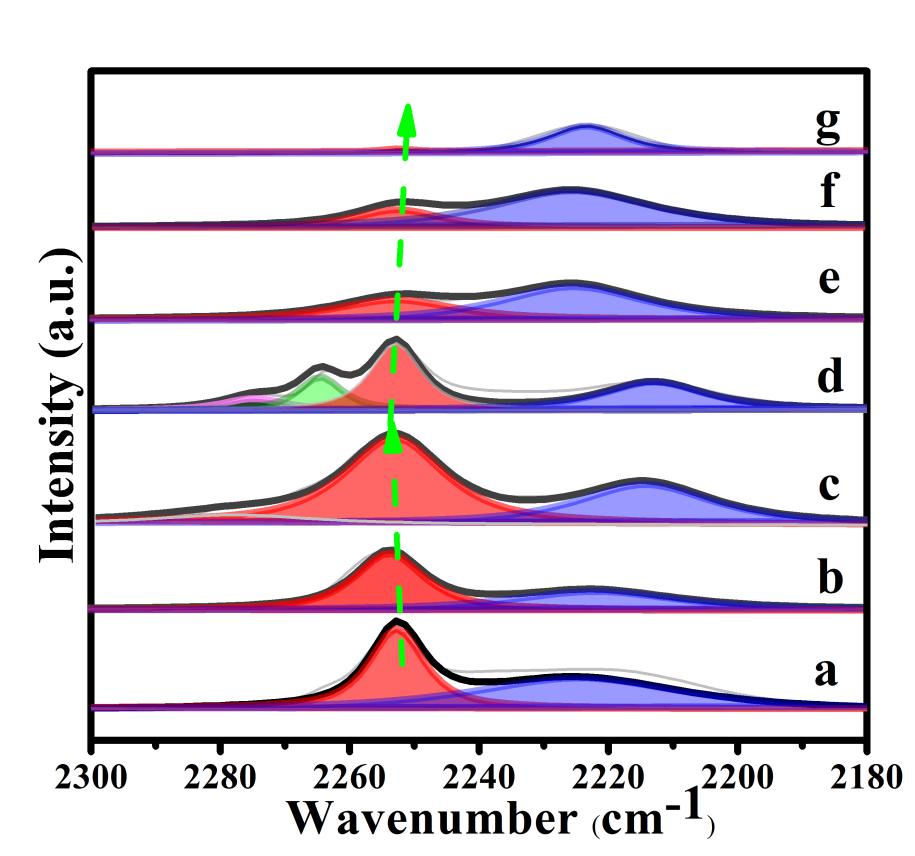
图3. 25 ℃下,七种样品在 2180−2300 cm-1对氘代氯仿分子的吸附图; (a) re-Ca4Al-LDHs, (b) re-Ca4Al0.95Ga0.05-LDHs, (c) re-Ca4Al0.90Ga0.10-LDHs, (d) re-Ca4Al0.85Ga0.15-LDHs, (e) re-Ca4Al0.95In0.05-LDHs, (f) re-Ca4Al0.90In0.10-LDHs, (g) re-Ca4Al0.85In0.15-LDHs。
构效关联
为了进一步探讨系列再水合重构样品的催化性能和碱性位点浓度之间的关系,以及Ga、In三价金属离子修饰对其催化活性的提升机制,我们将re-Ca4Al1-xGax-LDHs和re-Ca4Al1-xInx-LDHs样品的HPA初始生成速率与弱碱性和中强碱性位点的相对浓度进行线性关联,结果分别如图3-3A和3B所示。在图3-3A中可以观察到,HPA的初始生成速率与弱碱性位点的相对浓度呈现良好的正相关关系,说明弱碱性位点是该反应的催化活性位点。此外,结果还表明随着Ga 掺杂量的提升,re-Ca4Al1-xGax-LDHs中弱碱性位点的数量也有所增加,因此该类催化剂较re-Ca4Al-LDHs展现出更优异的催化活性。与之所不同的是,随着re-Ca4Al1-xInx-LDHs中In含量的增加,其弱碱性位点的比例明显降低,这与其较差的羟醛缩合反应活性有直接关系。
表1不同催化剂样品对甲醛与异丁醛反应的性能
Sample | Proton affinitya (KJmol-1) | BW/BW+BMb (area g-1) | BM/BW+BMb (area g-1) | IBD Conversion (%) | HPA Yield (%) | Formation Rate(mmol g-1 h-1)c |
re-Ca4Al | 838 | 0.55 | 0.44 | 80.3 | 60.5 | 57.1 |
re-Ca4Al0.95Ga0.05 | 830 | 0.58 | 0.42 | 82.3 | 62.4 | 73.3 |
re-Ca4Al0.90Ga0.10 | 827 | 0.63 | 0.36 | 91.0 | 71.8 | 104 |
re-Ca4Al0.85Ga0.15 | 828 | 0.59 | 0.41 | 88.5 | 68.1 | 88.0 |
re-Ca4Al0.95In0.05 | 842 | 0.30 | 0.65 | 77.0 | 58.1 | 34.5 |
re-Ca4Al0.90In0.10 | 843 | 0.20 | 0.79 | 70.5 | 50.0 | 24.9 |
re-Ca4Al0.85In0.15 | 845 | 0.05 | 0.95 | 68.0 | 48.6 | 16.7 |
a通过公式一计算出来的质子结合能。bBW和BM分别根据CDCl3在弱碱性位点和中碱性位点上的吸收峰面积除以样品量计算所得。cHPA初始生成速率由IBD转化率为2%时,对产物与反应时间曲线的切线的斜率进行计算所得。
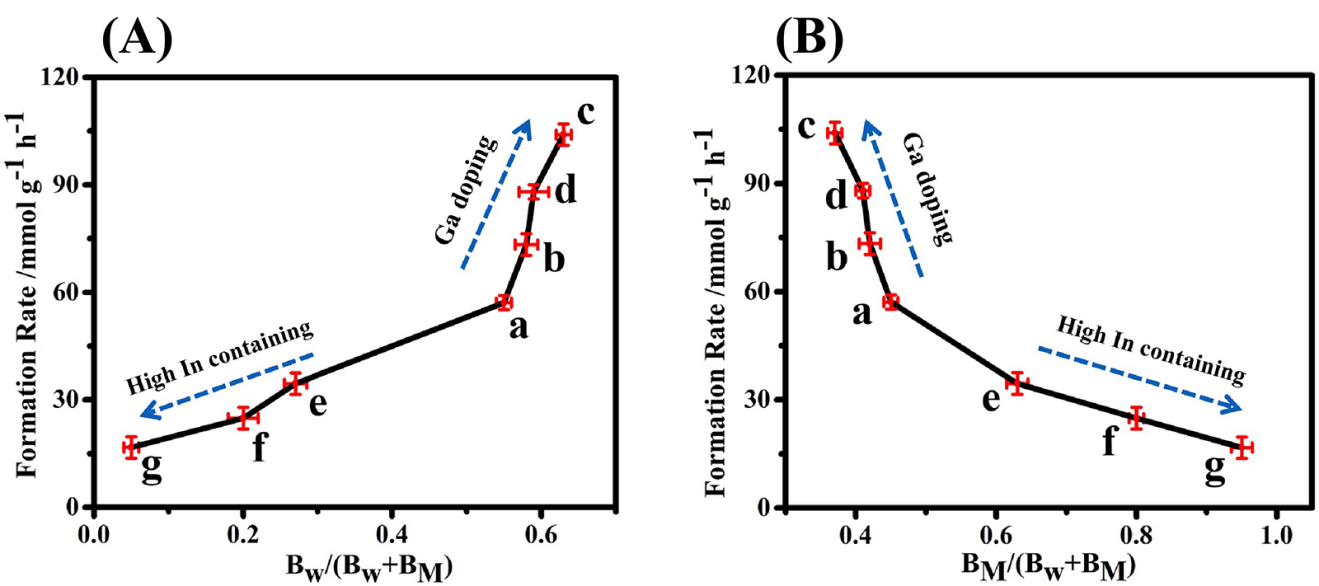
图4. 七种催化剂样品HPA初始生成速率与弱碱性位点与中强碱位点的相对浓度之间的函数关系。
三价金属M的掺杂与催化剂结构关联研究
为进一步得到三价金属M的掺杂与催化剂结构关联在原子层面的具体信息,通过XANES和EXAFS原位实验表征手段,对re-Ca4Al-LDHs、re-Ca4Al0.90Ga0.10-LDHs 和re-Ca4Al0.90In0.10-LDHs 样品中Ca2+的配位结构进行了详细的研究。图4A是参照样Ca(OH)2以及三种催化剂的Ca K边 XANES谱图。如图所示,与Ca(OH)2样品相比,三种催化剂样品的XANES吸收曲线明显不同,其在边前约4043 eV位置处出现了一个强度较弱的吸收峰,表明re-Ca4Al1-xMx-LDHs(M=Ga、In) 与Ca(OH)2相比,其Ca-OH的配位结构明显不同于对照样中的6配位结构。根据电子跃迁规则可对其原因进行推测,1s电子跃迁频率的降低可能与re-Ca4Al1-xMx-LDHs水滑石体系中Ca-OH正八面体的结构畸变有关,Ca2+具有相对较大的离子半径,当其在形成水滑石的晶格结构时容易发生畸变导致水滑石正八面体结构的对称性下降,从而使得层板中的Ca2+更容易与层间水分子形成非中心对称的7配位结构。
此外,根据Ca K边EXAFS谱图(4B), 可以看出在1.8 Å和3.0 Å处三种水滑石材料的R空间曲线都出现了相应的吸收峰,这两个吸收峰分别对应于Ca-O键与Ca-O-Ca键。当在钙铝水滑石中掺入Ga和In后,其Ca-O键的键长均发生了明显变化,其中,Ga的掺杂使得Ca-O键键长由1.719 Å增长到1.769 Å,而掺入In后,Ca-O键键长进一步增加到1.781 Å,表明Ga和In的掺入使得钙铝水滑石的晶格发生膨胀。比较有意思的是当掺入镓元素后,Ca-OH第一壳层的配位数有所增大,而掺入In后,Ca-OH第一壳层的配位数相较于钙铝水滑石却有所降低,这可能与两种掺杂元素所造成的羟醛缩合催化活性的差异有直接关系。
为了进一步探索这一不寻常的现象,我们利用In(OH)3作为参照样品,通过In L边EXAFS和In 3d XPS表征来详细考察re-Ca4Al1-xInx-LDHs体系中In的配位状态,从图4C中可以看出,与In(OH)3相比,re-Ca4Al0.90In0.10-LDHs样品中In-OH的配位数明显增加,说明在re-Ca4Al0.90In0.10-LDHs中存在高度配位的In-OH基团。因此,根据EXAFS谱图和催化评价的结果,认为Ga作为结构促进剂掺入到水滑石层板中会引起适度的晶格膨胀,从而导致水滑石层板发生一定程度的畸变和暴露更多的活性位点。这与FTIR的结果一致,即Ga的引入导致了弱碱性位点(7配位Ca-OH)数量的显著增加,其在很大程度上促进了IBD的转化。相较而言,随着re-Ca4Al0.90In0.10-LDHs体系中In含量的增加,其IBD转化率明显降低。根据Ca K边和In L边EXAFS分析结果可以得出,将具有较大离子半径的In掺杂进水滑石层板中会造成水滑石层板结构的显著扩张,导致主体层板发生畸变,使得层板中的In易与层间水分子结合形成高配位的In-OH位点。这时In3+的存在与层板中的Ca2+形成一种竞争的关系,导致7配位的Ca-OH活性位点的数量减少,这与CDCl3-FTIR和催化评价结果相一致。
为进一步探究三种催化剂中氧物种的结构,对re-Ca4Al0.90Ga0.10-LDHs、re-Ca4Al0.90In0.10-LDHs和re-Ca4Al-LDHs样品的O l s XPS谱图(图4D)进行了测定。对于三种样品来说, 有一个宽泛且不对称的O 1s吸收峰出现在529 eV536 eV之间。根据相关文献报道和课题组前期工作研究,将该O 1s宽峰进行高斯-洛伦兹分峰拟合可将其分为三个峰,其中,在532.4 eV,531.4 eV 和 530.4 eV左右位置处的峰可分别归属于7配位羟基氧,晶格氧和低配位氧的峰。从图4D可以看出,这些样品中七配位羟基氧的结合能差异并不明显(在532.5 eV和532.3 eV之间),说明Ga或In的掺入对re-LDHs催化剂中氧的电子密度影响不大,并没有引起碱性强度的显著变化,与CDCl3-FTIR结果一致。
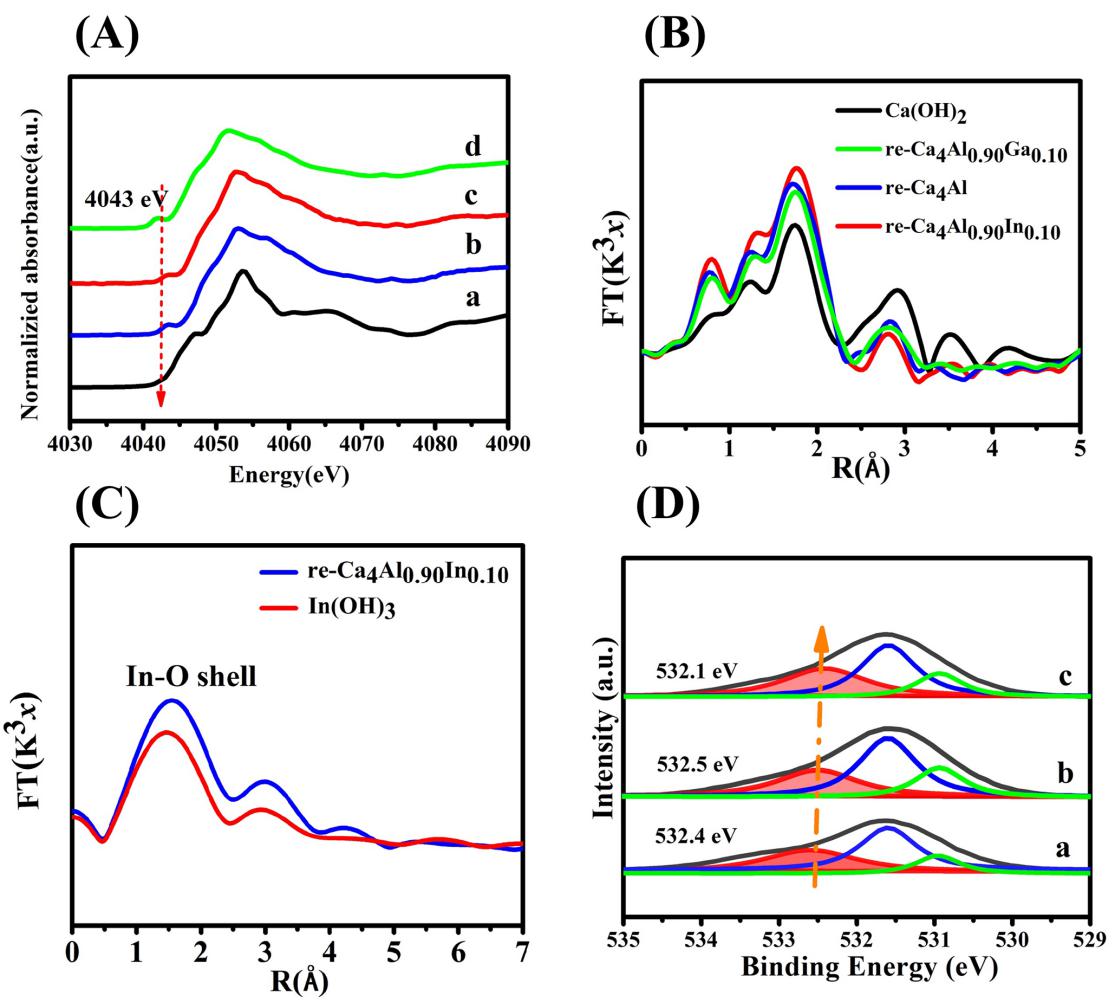
图5.在四种样品上的谱图(a) Ca(OH)2, (b) re-Ca4Al-LDHs, (c) re-Ca4Al0.90Ga0.10-LDHs和(d) re-Ca4Al0.90In0.10-LDHs (A)Ca K近边吸收曲线 (B) Ca K边EXAFS (C) In L边EXAFS (D) In 3d XPS。
全文小结:
该论文围绕一种新型高效Ca基固体碱催化剂的制备及性能调控,用于替代现阶段工业制备新戊二醇所采用的液体碱催化剂展开了相关研究。基于LDHs前体材料的主体组成可调变性和结构记忆效应,经元素掺杂及水合复原过程设计和制备了系列re-Ca4Al1-xMx-LDHs(M=Ga、In)催化剂, 研究了其对甲醛与异丁醛羟醛缩合制备新戊二醇的关键中间体-羟基新戊醛的催化性能。借助XRD、SEM和N2-吸脱附等表征手段,探究了具有不同离子半径的元素掺杂对re-Ca4Al1-xMx-LDHs结构性质的影响规律。通过对反应温度、反应气氛和反应时间进行系统优化,在保证高转化率和选择性的前提下获得了对目标产物HPA的高收率。其中,该催化能力明显高于其它同类固体碱催化剂,与工业上经羟醛缩合反应制备HPA所采用的液体碱催化剂的水平相当。利用X射线精细结构吸收谱、氘代氯仿原位红外等结构表征手段及密度泛函理论计算,深入研究了结构助剂Ga和In的掺杂对LDHs固体碱催化剂碱性位点的调控规律,揭示了碱性位结构对羟醛缩合反应的催化作用机制。本论文为新型Ca基固体碱催化剂的结构设计及性能强化提供了新思路,具有一定的理论价值及工业应用前景。
作者介绍:
第一作者:王慧敏,女,硕士研究生,北京化工大学。2013年考入安阳师范学院化学专业,2017年获得安阳师范学院理学学士学位。同年进入北京化工大学理学院化学专业,攻读硕士学位。专业为化学,师从卫敏教授,研究方向为水滑石基固体碱催化剂的碱性位结构调控及其对羟醛缩合反应的性能研究。
通讯作者:
文章信息:
Huimin Wang, Weihan Bing, Chunyuan Chen, Yusen Yang, Ming Xu, Lifang Chen, Lei Zheng, Xiaolin Li, Xin Zhang *, Jianjun Yin *, Min Wei *,Chin. J. Catal., 2020, 41: 1279–1287.