
原子有序双金属Ni-Mo催化剂实现低压下糠醛高效催化转化

1、全文速览:
近年来,富氧生物质催化转化为烃类化合物在能源与环境催化领域引起了极大关注,其中糠醛催化加氢脱氧(HDO)制2-甲基呋喃(2-MF)是一个典型的范例。然而,由于该过程涉及C=O键的选择性加氢以及随后C-OH键的裂解等复杂步骤,因此如何在温和条件下实现对目标产物的单一选择性仍然是一个巨大的挑战。近日,我课题组报道了一种由层状前体拓扑法制备的NiMo金属间化合物(Intermetallic Compound, IMC)催化剂,其在较低的氢压(0.1 MPa)下对糠醛加氢脱氧制2-MF的反应表现出优异的催化性能(收率:99%)。通过AC-STEM,XAFS等结构表征以及原位FT-IR与DFT计算相结合的研究手段,证明了IMC中原子有序的活性位点对于催化反应的决定性作用。
2、背景介绍:
石油资源的枯竭和环境问题的加剧迫使人类寻找可再生的新能源,其中生物质基化学品和燃料是发展可持续化学工业的一个很有前景的机会。糠醛是一种由木糖脱水水解得到的重要生物质平台分子,通过加氢、氢解等过程可将其转化为一系列高附加值的燃料和化学品。特别是糠醛加氢脱氧制得的2-MF在汽油添加剂、关键溶剂、香料前体和医药中间体等领域的广泛应用,使其引起了工业和学术界的广泛关注。该反应的难点在于需要对C=O键进行选择性加氢,随后对C-OH键进行裂解,同时保持呋喃环内的C-O/C-C/C=C键完整。鉴于单金属催化剂在催化活性和选择性上的不足,前人的研究工作致力于双金属催化剂的设计与制备以在高活性的前提下获得高产率的2-MF。然而,对于新型催化剂的合理设计、催化活性中心的精准调控以及对双金属催化剂的构效关联的深入认识还很欠缺。此外,在液相反应体系中,该反应通常需要较高的氢气压力(通常在1 MPa以上),这会导致严重的安全、设备和使用效率问题,从而限制了其实际应用。
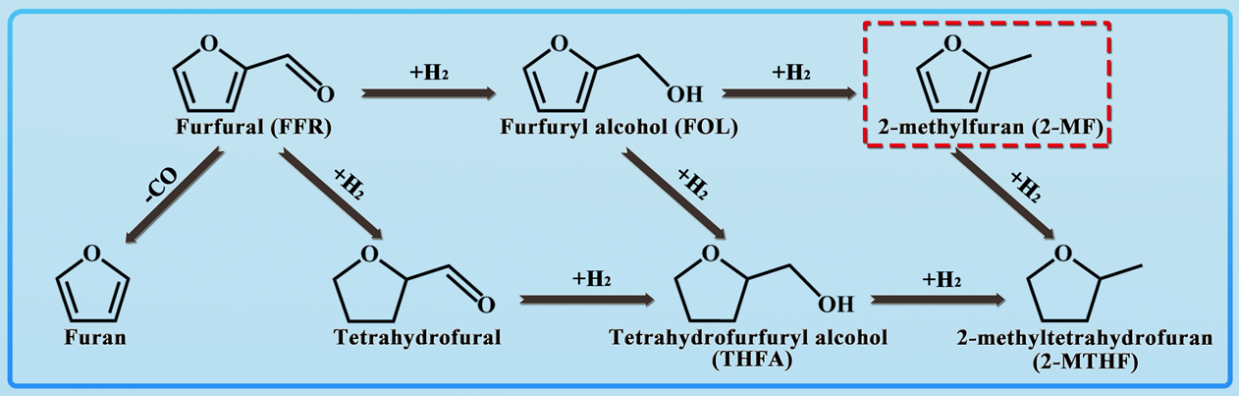
图1 糠醛加氢脱氧反应路径图。
3、研究出发点:
针对以上问题和挑战,作者以镍铝水滑石(NiAl-LDHs)作为前体外源引入亲氧性较强的Mo元素,基于层状前体拓扑转变过程制备了具有原子有序活性位点的NiMo金属间化合物催化剂,实现了在较低的氢压(0.1 MPa)下糠醛高效加氢脱氧制2-MF(收率99%)。结合结构表征,原位实验与理论计算等手段,系统研究了双金属催化剂的原子结构与底物的活化吸附、转化路径以及产物选择性的关系,揭示了原子有序的Ni-Mo活性位点在反应中的决定性作用,为合理设计和开发高效的加氢脱氧催化剂提供了一定的理论指导。
4、图文解析:
催化剂制备与结构表征:
采用原位生长法合成了花状NiAl-LDHs前体,结合等体积浸渍法外源引入Mo元素,调控层状前体的结构拓扑转变过程,制备了Al2O3负载的单金属Ni、NiMo合金和NiMo IMC样品。TEM显示三个样品的粒径分别为10.4 nm、13.9 nm和18.6 nm,HRTEM观察到相应的晶格条纹:Ni(d111 = 0.203 nm),NiMo alloy(d111 = 0.205 nm)和NiMo IMC(d331 = 0.209 nm)。NiMo alloy的AC-HAADF-STEM图像(图2D1)显示,晶格中的Ni原子被Mo原子随机取代(Mo的亮度高于Ni),相应的EDS mapping图像(图2C1)显示Ni和Mo富集在单个纳米粒子的不同区域中,表明元素分布不均,这进一步证实了无序合金的形成。对于NiMo IMC样品,EDS mapping图像显示Ni和Mo均匀分散在一个纳米颗粒中(图2C2)。 高倍率HAADF-STEM图像(图2D2)显示了Ni和Mo原子的交替有序排列,其晶格间距为2.09 Å,对应于NiMo IMC相的(331)面。反FFT模式(图2E2)和相应的线扫曲线(图2F2)进一步证明了这种高度有序的原子结构。
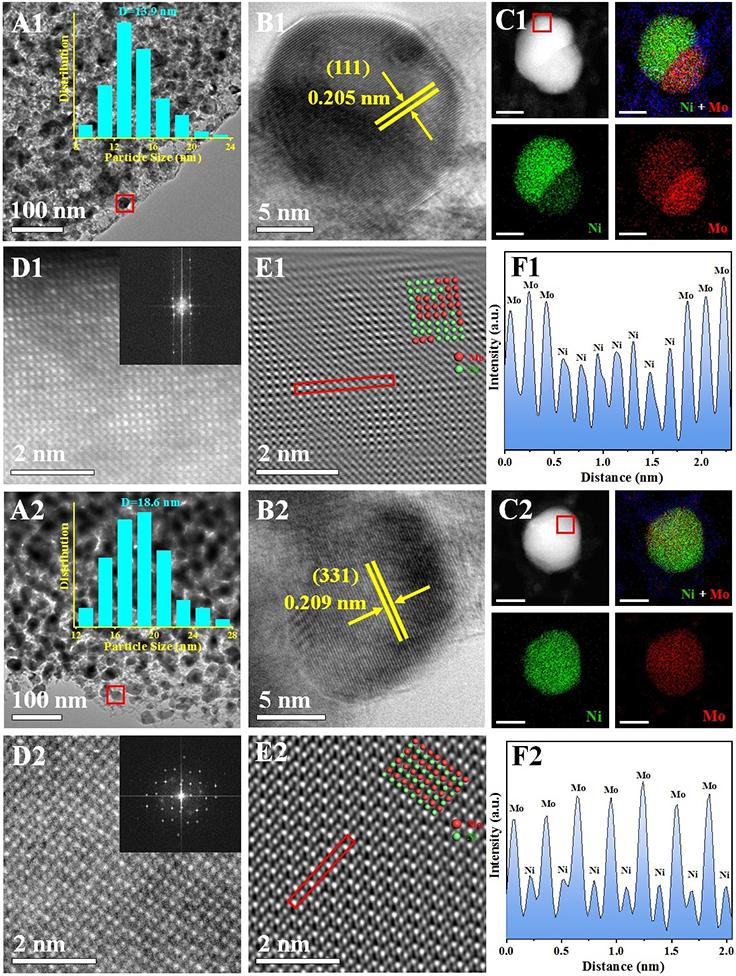
图2 NiMo alloy (A1-F1)和NiMo IMC (A2-F2)的TEM、HRTEM图像、元素分布图以及相应的HAADF-STEM图像和线扫曲线。
XPS结果表明(图3A和B),随着Mo原子的加入,Ni0峰值(单金属Ni为852.8 eV)在NiMo alloy中移至852.5 eV,在NiMo IMC中进一步移至852.3 eV,而Mo0峰值则向高结合能移动,说明Ni在NiMo alloy和NiMo IMC中的电负性增强,存在由Mo向Ni的电子偏移。CO吸附原位红外光谱(图3C)、Bader电荷分析以及XANES实验结果进一步证明了这一结论。原位EXAFS实验(图3D和E)发现,相比于单金属Ni样品,NiMo alloy和NiMo IMC的Ni-Ni键键长逐渐增加,Ni-Ni配位数逐渐降低,NiMo alloy样品的Ni-Mo配位数约为3,而NiMo IMC则增加至5,进一步证明了原子有序的IMC与无序alloy之间的结构差异。
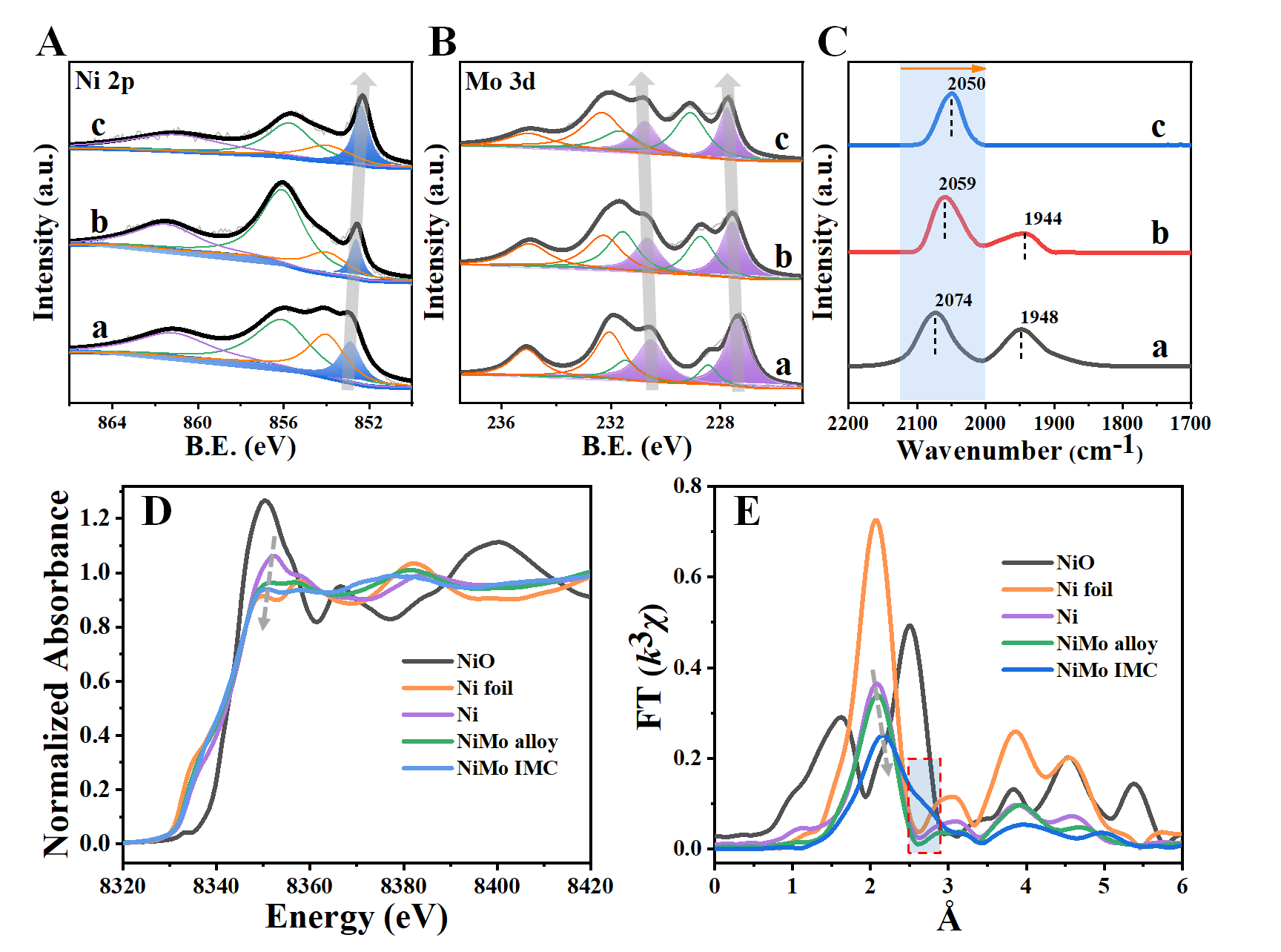
图3 样品的Ni 2p轨道(A)和Mo 3d轨道 (B)的XPS谱图,金属Ni、NiMo alloy和NiMo IMC上CO吸附的原位FT-IR光谱(C),Ni K边归一化的XANES光谱 (D)以及傅里叶变换k3加权的EXAFS光谱 (E)。
催化剂的糠醛加氢脱氧性能评价:
在较低的氢气压力(0.1 MPa)下,研究了不同催化剂对糠醛加氢脱氧制2-MF的催化性能。如图4A所示,所有的Ni基催化剂在4 h后都表现出100%的糠醛转化率,并且单金属Ni的反应速率高于NiMo alloy和NiMo IMC,但是对2-MF的选择性(图4B–D)差异很大。在单金属Ni样品上(图4B)糠醛分子中的C=O和C=C均被氢化,对四氢呋喃(THFA)的选择性为82%,但是它活化C-OH的能力很弱,2-MF的选择性仅为9%。对于NiMo alloy(图4C),获得的2-MF选择性为37%,2-甲基四氢呋喃(2-MTHF)选择性为18%,表明对C-OH键的活化能力增强,但呋喃环加氢仍不可避免。令人惊讶的是,NiMo IMC催化剂(图4D)显示出对C=C加氢的完全抑制,同时C-OH基团发生了强烈的氢解反应,实现了对2-MF的单一选择性(99%),这优于文献报道的Ni基样品,甚至可与贵金属催化剂水平相当。并且,催化剂的循环稳定性良好。为了进一步验证中间产物糠醇(FOL)和反应途径,直接使用FOL作为反应物评价了催化剂性能,其2-MF产率与上述结果相近,进一步证明了NiMo IMC对C-OH基团突出的氢解作用。
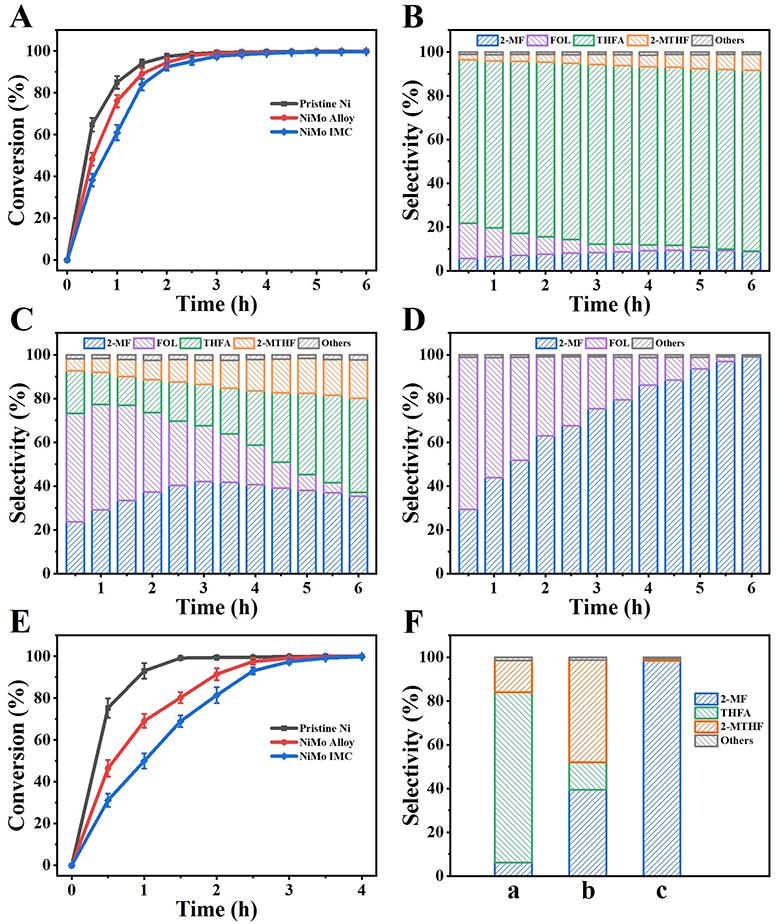
图4 糠醛的转化率(A)以及Ni (B)、NiMo alloy (C)和NiMo IMC (D)上产物分布随反应时间变化图;糠醇的转化率(E)以及不同催化剂上产物分布图(F)。
原位实验与理论计算相结合揭示构效关联:
通过原位FT-IR光谱研究了糠醛在催化剂表面的吸附和加氢过程。在单金属Ni表面C=O键和呋喃环均发生化学吸附(平行吸附构型);相比单金属Ni, 原子无序的NiMo alloy对于C=O键的吸附增强,而呋喃环的吸附略微减弱;而对于原子有序的NiMo IMC,其对于C=O的吸附显著增强,呋喃环的吸附则受到极大抑制。通入H2后,吸附于单金属Ni表面的糠醛分子优先进行C=C键的加氢,然后发生C=O加氢形成C-OH;类似地,NiMo alloy表面的也优先发生C=C键的加氢;而在IMC表面C=O的红外振动峰率先消失,C=C键的峰则下降缓慢,表明优先发生C=O的加氢,而生成的C-OH峰快速消失,这可能表明C-OH被进一步活化发生后续反应。DFT计算进一步表明,在NiMo IMC表面糠醛分子则以特定的倾斜构型吸附从而显著增强C=O的吸附,抑制呋喃环的加氢。糠醇(FOL)的原位FT-IR实验及DFT计算结果表明,NiMo IMC极大增强了糠醇C-OH键的吸附活化,促使其进一步发生氢解作用生成2-甲基呋喃。
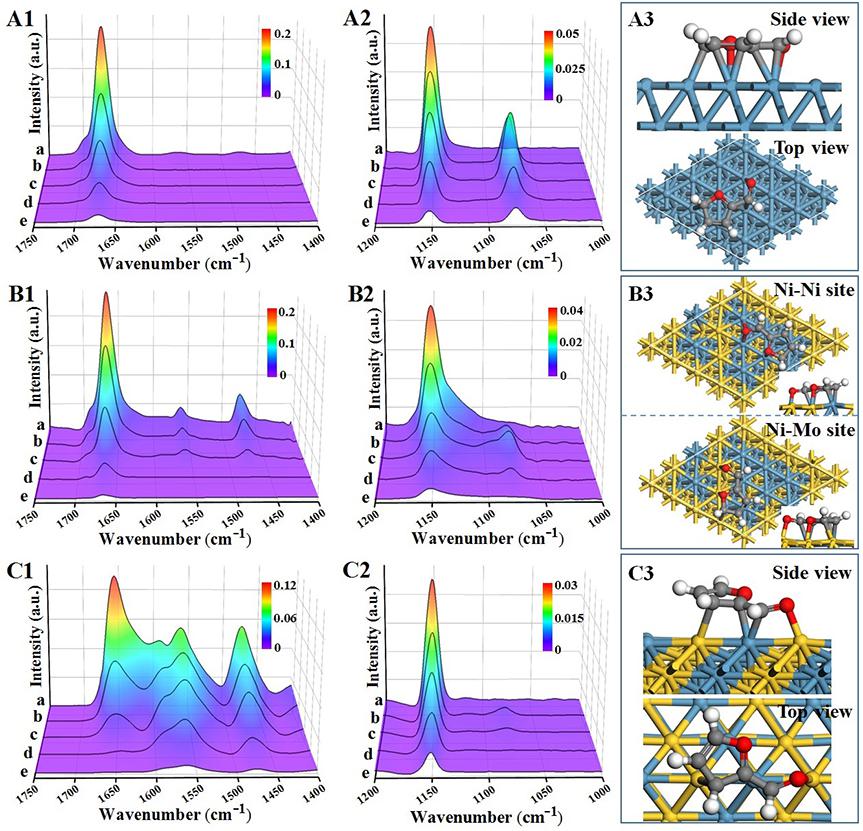
图5 Ni (A)、NiMo alloy (B)和NiMo IMC (C)上糠醛的吸附和加氢过程的原位FT-IR光谱以及DFT计算糠醛在催化剂表面的吸附构型。
FOL作为重要的中间产物,其转化路径决定了糠醛加氢脱氧反应的最终产物。基于上述吸附结构,通过DFT研究了FOL在Ni(111)和NiMo IMC(331)表面可能的加氢路径,并计算了其C-OH键断裂和呋喃环(C=C键)加氢的势垒。对于NiMo IMC,其C-OH键断裂能垒相比于单金属Ni显著降低,而C=C键加氢能垒显著升高;此外,Ni表面的C=C键加氢能势垒远低于C-OH键断裂势垒;而NiMo IMC上C=C键加氢的能垒要高于C-OH键断裂的能垒。这说明NiMo IMC表面的原子有序位点上,FOL分子优先经历C-OH键的断离,而抑制C=C键的加氢,从而单一选择性地产生2-MF。
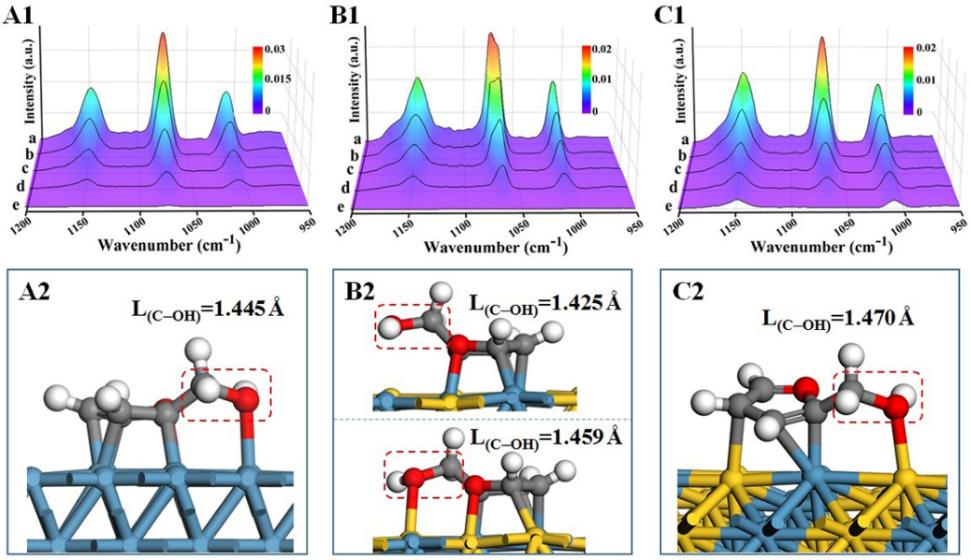
图6 糠醇在Ni (A)、NiMo alloy (B)和NiMo IMC (C)上吸附和反应过程的原位FT-IR光谱以及不同催化剂表面糠醇的吸附构型与C-OH键长。
综上,我们得出原子有序NiMo位点上糠醛加氢脱氧生成2-MF的反应路径:(1) 糠醛分子的C=O键被NiMo位点通过Mo-O键和Ni-C键活化吸附,呋喃环呈弱倾斜吸附;同时,氢在邻近的Ni原子上解离,形成活性氢原子;(2) C=O键发生加氢生成中间产物(FOL);(3) FOL的C-OH键随后被有序的NiMo位点裂解形成亚甲基呋喃和羟基;(4) 亚甲基呋喃和羟基与活性氢原子结合生成2-MF和H2O;(5) 2-MF分子从催化剂表面脱附。
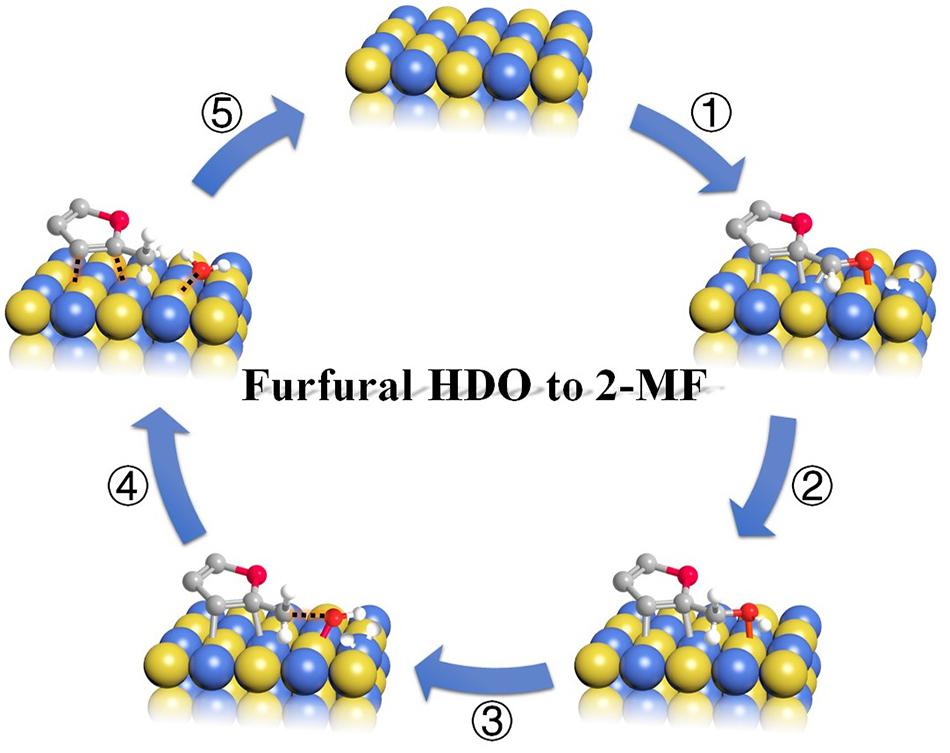
图7 NiMo IMC表面糠醛加氢脱氧生成2-MF的反应机理示意图。
5、总结与展望:
本工作基于层状前体拓扑法制备了NiMo IMC催化剂,其在较低的氢气压力(0.1 MPa)下对糠醛加氢脱氧转化为2-MF具有良好的催化性能(产率:99%),显著优于单金属Ni(9%),NiMo alloy(37%)和其他先前报道的Ni基催化剂。CO-IR,STEM,XAFS等结果证明,NiMo IMC中的Ni/Mo位点具有长程有序的原子序列,这与NiMo alloy中的随机原子排列形成鲜明对比。原位FT-IR和DFT计算表明,在NiMo IMC表面,只有糠醛的C=O在Ni-Mo双金属活性位上发生桥式吸附,而呋喃环的吸附受到极大抑制。而且,原子有序的Ni-Mo位点降低了FOL中C-OH键的活化裂解能垒,从而生成2-MF。NiMo IMC催化剂具有在相对较低的操作压力下对目标产物高收率的优势,具有潜在的应用前景。
6、作者介绍:
第一作者:
刘巍,男,博士研究生。2016年于东北林业大学获得工学学士学位。2017年进入北京化工大学理学院化学工程与技术专业,攻读硕士学位,师从卫敏教授,2019年转为硕博连读。主要研究方向为双金属催化剂的原子尺度活性位调控及其在加氢脱氧反应中的应用。